Publications
2025
-
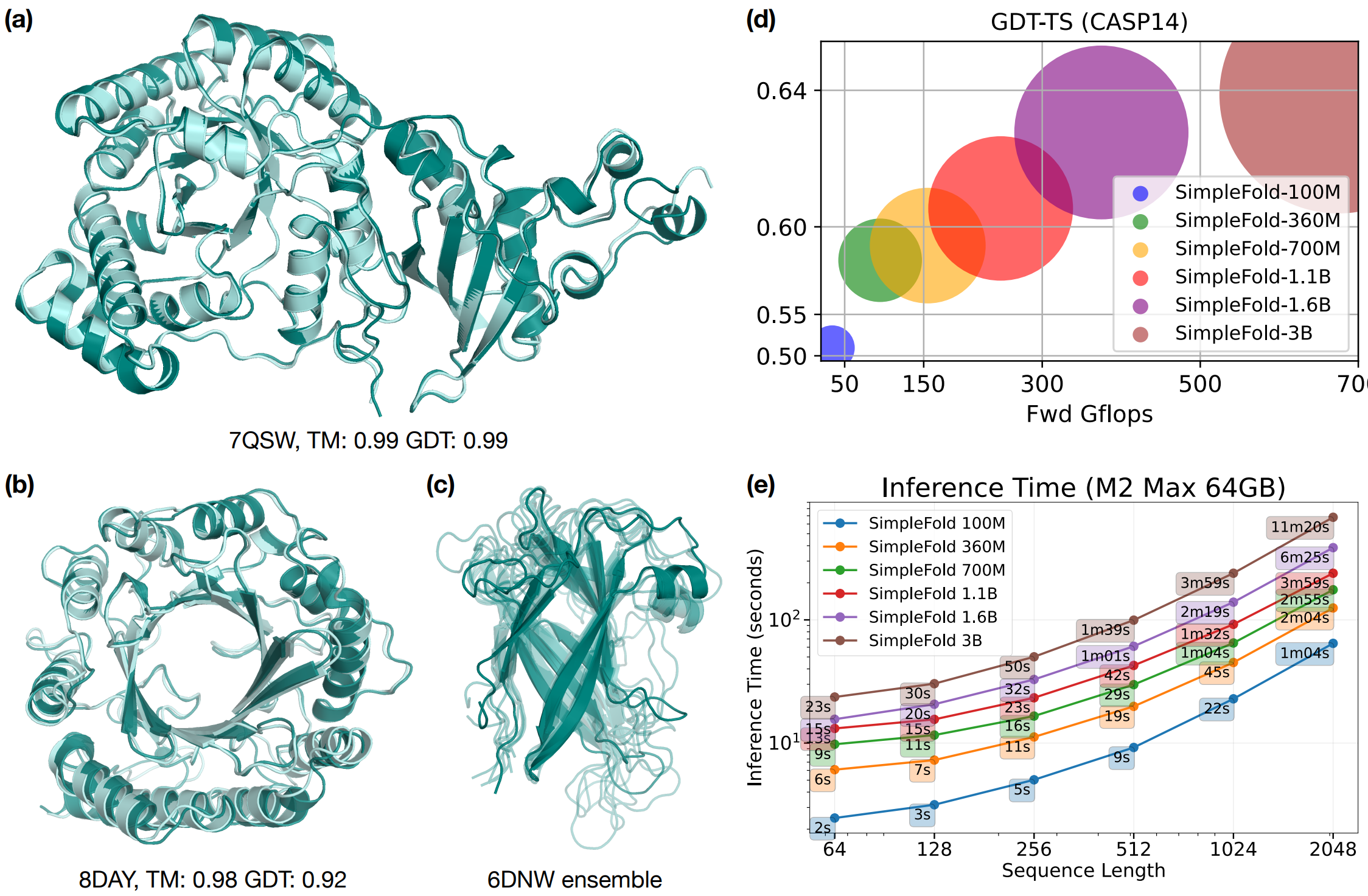 SimpleFold: Folding Proteins is Simpler than You ThinkYuyang Wang, Jiarui Lu, Navdeep Jaitly, Josh Susskind, and Miguel Angel BautistaarXiv preprint arXiv:2509.18480, 2025
SimpleFold: Folding Proteins is Simpler than You ThinkYuyang Wang, Jiarui Lu, Navdeep Jaitly, Josh Susskind, and Miguel Angel BautistaarXiv preprint arXiv:2509.18480, 2025Protein folding models have achieved groundbreaking results typically via a combination of integrating domain knowledge into the architectural blocks and training pipelines. Nonetheless, given the success of generative models across different but related problems, it is natural to question whether these architectural designs are a necessary condition to build performant models. In this paper, we introduce SimpleFold, the first flow-matching based protein folding model that solely uses general purpose transformer blocks. Protein folding models typically employ computationally expensive modules involving triangular updates, explicit pair representations or multiple training objectives curated for this specific domain. Instead, SimpleFold employs standard transformer blocks with adaptive layers and is trained via a generative flow-matching objective with an additional structural term. We scale SimpleFold to 3B parameters and train it on approximately 9M distilled protein structures together with experimental PDB data. On standard folding benchmarks, SimpleFold-3B achieves competitive performance compared to state-of-the-art baselines, in addition SimpleFold demonstrates strong performance in ensemble prediction which is typically difficult for models trained via deterministic reconstruction objectives. Due to its general-purpose architecture, SimpleFold shows efficiency in deployment and inference on consumer-level hardware. SimpleFold challenges the reliance on complex domain-specific architectures designs in protein folding, opening up an alternative design space for future progress.
-
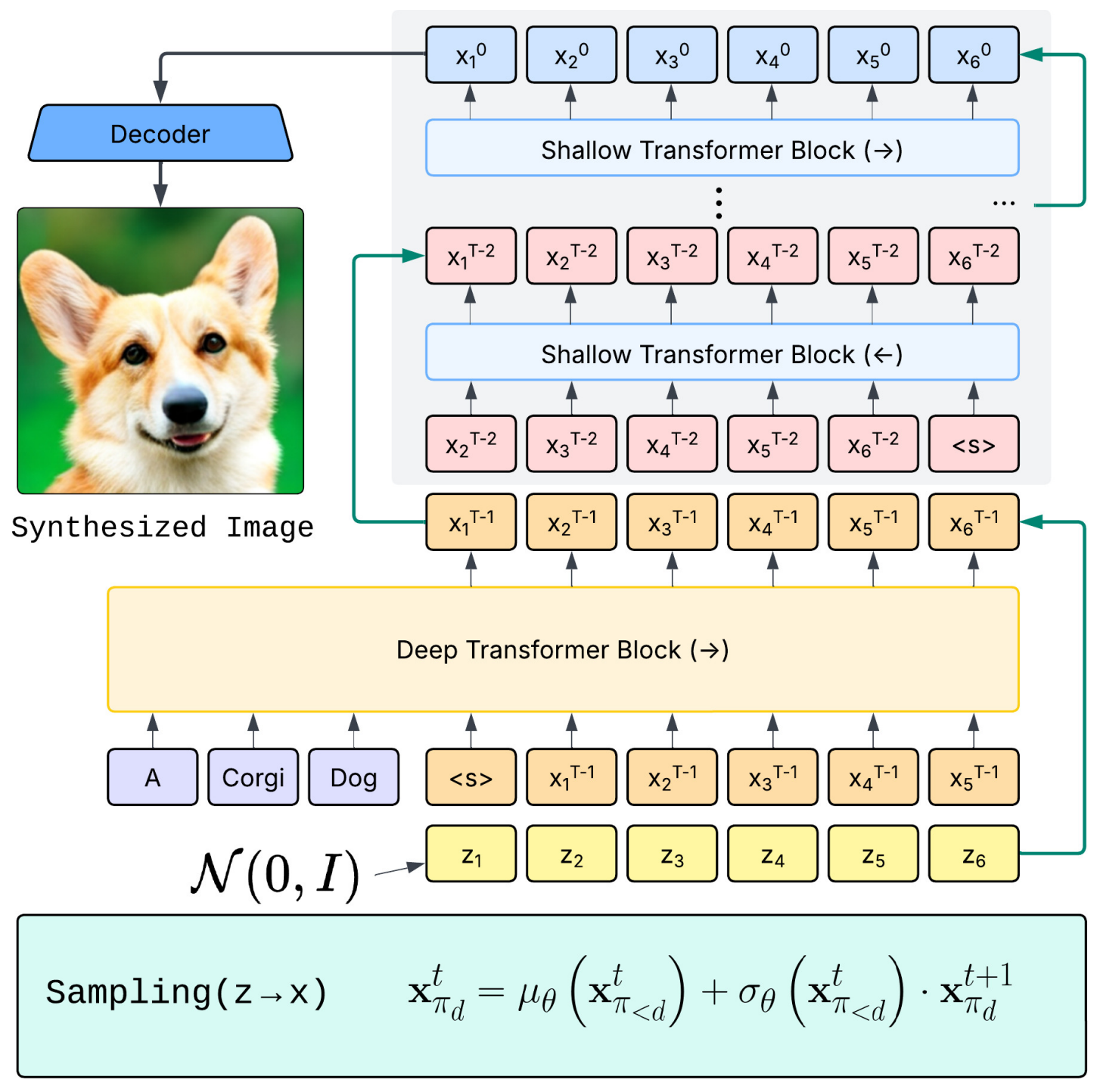 STARFlow: Scaling Latent Normalizing Flows for High-resolution Image SynthesisJiatao Gu, Tianrong Chen, David Berthelot, Huangjie Zheng, Yuyang Wang, Ruixiang Zhang, Laurent Dinh, Miguel Angel Bautista, Josh Susskind, and Shuangfei ZhaiarXiv preprint arXiv:2506.06276, 2025
STARFlow: Scaling Latent Normalizing Flows for High-resolution Image SynthesisJiatao Gu, Tianrong Chen, David Berthelot, Huangjie Zheng, Yuyang Wang, Ruixiang Zhang, Laurent Dinh, Miguel Angel Bautista, Josh Susskind, and Shuangfei ZhaiarXiv preprint arXiv:2506.06276, 2025We present STARFlow, a scalable generative model based on normalizing flows that achieves strong performance in high-resolution image synthesis. The core of STARFlow is Transformer Autoregressive Flow (TARFlow), which combines the expressive power of normalizing flows with the structured modeling capabilities of Autoregressive Transformers. We first establish the theoretical universality of TARFlow for modeling continuous distributions. Building on this foundation, we introduce several key architectural and algorithmic innovations to significantly enhance scalability: (1) a deep-shallow design, wherein a deep Transformer block captures most of the model representational capacity, complemented by a few shallow Transformer blocks that are computationally efficient yet substantially beneficial; (2) modeling in the latent space of pretrained autoencoders, which proves more effective than direct pixel-level modeling; and (3) a novel guidance algorithm that significantly boosts sample quality. Crucially, our model remains an end-to-end normalizing flow, enabling exact maximum likelihood training in continuous spaces without discretization. STARFlow achieves competitive performance in both class-conditional and text-conditional image generation tasks, approaching state-of-the-art diffusion models in sample quality. To our knowledge, this work is the first successful demonstration of normalizing flows operating effectively at this scale and resolution.


2024
-
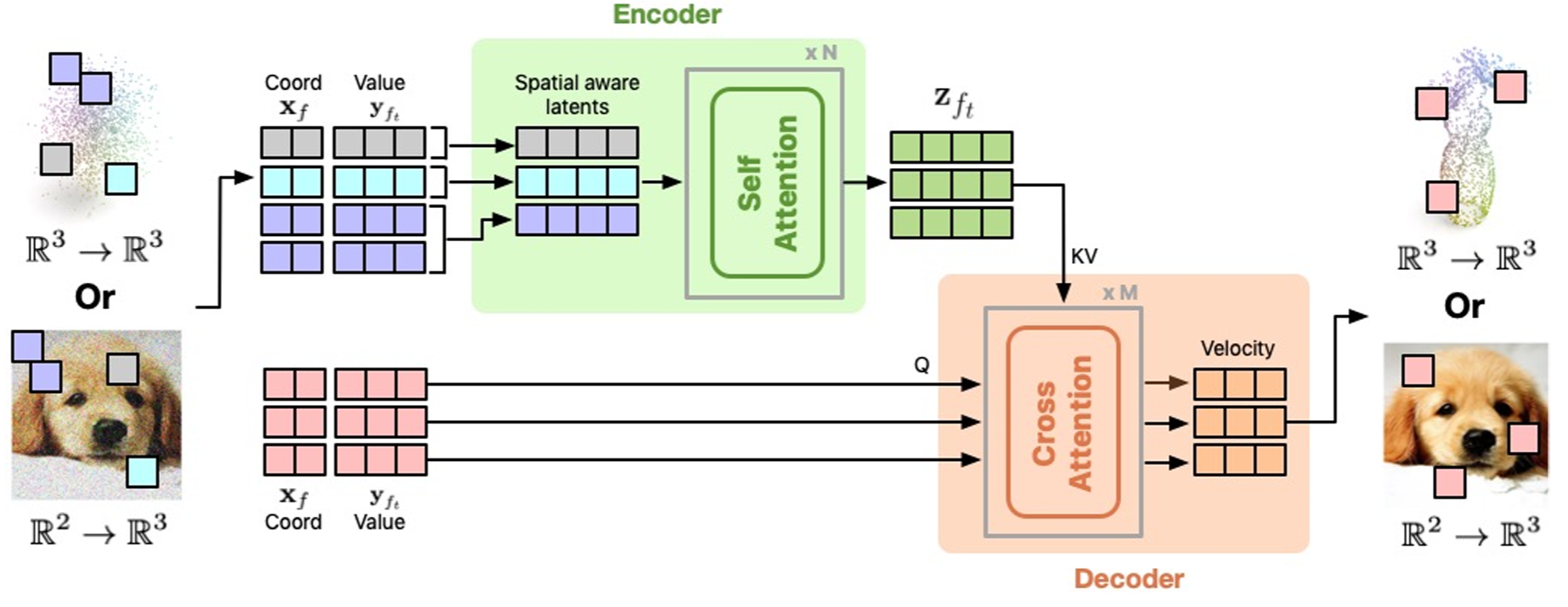 INRFlow: Flow Matching for INRs in Ambient SpaceYuyang Wang, Anurag Ranjan, Josh Susskind, and Miguel Angel BautistaThe International Conference on Machine Learning (ICML), 2024
INRFlow: Flow Matching for INRs in Ambient SpaceYuyang Wang, Anurag Ranjan, Josh Susskind, and Miguel Angel BautistaThe International Conference on Machine Learning (ICML), 2024Flow matching models have emerged as a powerful method for generative modeling on domains like images or videos, and even on irregular or unstructured data like 3D point clouds or even protein structures. These models are commonly trained in two stages: first, a data compressor is trained, and in a subsequent training stage a flow matching generative model is trained in the latent space of the data compressor. This two-stage paradigm sets obstacles for unifying models across data domains, as hand-crafted compressors architectures are used for different data modalities. To this end, we introduce INRFlow, a domain-agnostic approach to learn flow matching transformers directly in ambient space. Drawing inspiration from INRs, we introduce a conditionally independent point-wise training objective that enables INRFlow to make predictions continuously in coordinate space. Our empirical results demonstrate that INRFlow effectively handles different data modalities such as images, 3D point clouds and protein structure data, achieving strong performance in different domains and outperforming comparable approaches. INRFlow is a promising step towards domain-agnostic flow matching generative models that can be trivially adopted in different data domains.
-
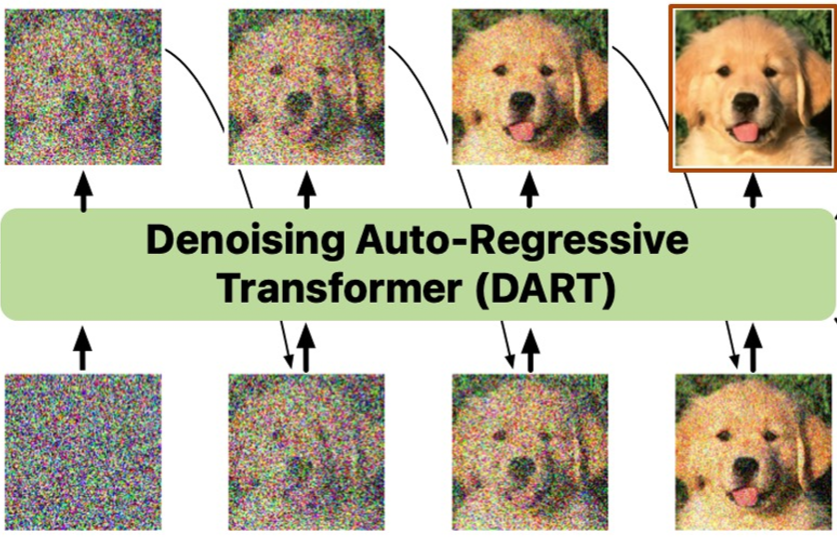 DART: Denoising autoregressive transformer for scalable text-to-image generationJiatao Gu, Yuyang Wang, Yizhe Zhang, Qihang Zhang, Dinghuai Zhang, Navdeep Jaitly, Josh Susskind, and Shuangfei ZhaiInternational Conference on Learning Representations (ICLR), 2024
DART: Denoising autoregressive transformer for scalable text-to-image generationJiatao Gu, Yuyang Wang, Yizhe Zhang, Qihang Zhang, Dinghuai Zhang, Navdeep Jaitly, Josh Susskind, and Shuangfei ZhaiInternational Conference on Learning Representations (ICLR), 2024Diffusion models have become the dominant approach for visual generation. They are trained by denoising a Markovian process that gradually adds noise to the input. We argue that the Markovian property limits the models ability to fully utilize the generation trajectory, leading to inefficiencies during training and inference. In this paper, we propose DART, a transformer-based model that unifies autoregressive (AR) and diffusion within a non-Markovian framework. DART iteratively denoises image patches spatially and spectrally using an AR model with the same architecture as standard language models. DART does not rely on image quantization, enabling more effective image modeling while maintaining flexibility. Furthermore, DART seamlessly trains with both text and image data in a unified model. Our approach demonstrates competitive performance on class-conditioned and text-to-image generation tasks, offering a scalable, efficient alternative to traditional diffusion models. Through this unified framework, DART sets a new benchmark for scalable, high-quality image synthesis.
-
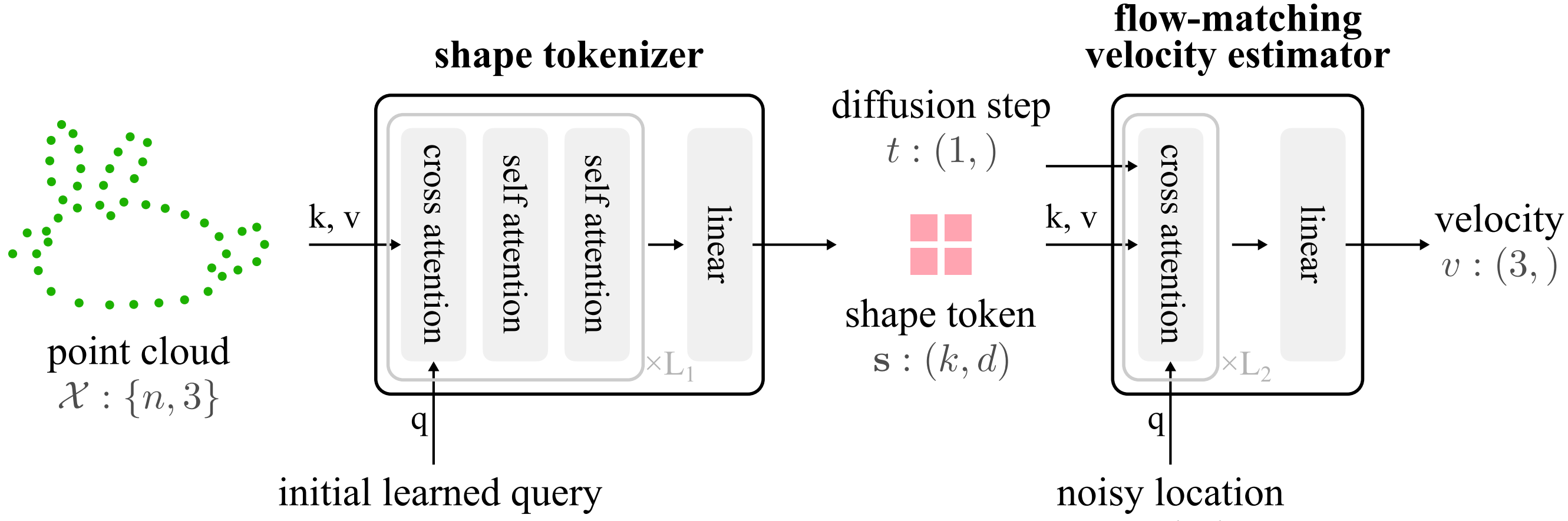 3D Shape TokenizationJen-Hao Rick Chang, Yuyang Wang, Miguel Angel Bautista Martin, Jiatao Gu, Josh Susskind, and Oncel TuzelarXiv preprint arXiv:2412.15618, 2024
3D Shape TokenizationJen-Hao Rick Chang, Yuyang Wang, Miguel Angel Bautista Martin, Jiatao Gu, Josh Susskind, and Oncel TuzelarXiv preprint arXiv:2412.15618, 2024We introduce Shape Tokens, a 3D representation that is continuous, compact, and easy to incorporate into machine learning models. Shape Tokens act as conditioning vectors that represent shape information in a 3D flow-matching model. The flow-matching model is trained to approximate probability density functions corresponding to delta functions concentrated on the surfaces of shapes in 3D. By attaching Shape Tokens to various machine learning models, we can generate new shapes, convert images to 3D, align 3D shapes with text and images, and render shapes directly at variable, user specified, resolution. Moreover, Shape Tokens enable a systematic analysis of geometric properties such as normal, density, and deformation field. Across all tasks and experiments, utilizing Shape Tokens demonstrate strong performance compared to existing baselines.
-
 Swallowing the Bitter Pill: Simplified Scalable Conformer GenerationYuyang Wang, Ahmed A. Elhag, Navdeep Jaitly, Joshua Susskind, and Miguel Angel BautistaInternational Conference on Machine Learning (ICML), 2024
Swallowing the Bitter Pill: Simplified Scalable Conformer GenerationYuyang Wang, Ahmed A. Elhag, Navdeep Jaitly, Joshua Susskind, and Miguel Angel BautistaInternational Conference on Machine Learning (ICML), 2024We present a novel way to predict molecular conformers through a simple formulation that sidesteps many of the heuristics of prior works and achieves state of the art results by using the advantages of scale. By training a diffusion generative model directly on 3D atomic positions without making assumptions about the explicit structure of molecules (e.g. modeling torsional angles) we are able to radically simplify structure learning, and make it trivial to scale up the model sizes. This model, called Molecular Conformer Fields (MCF), works by parameterizing conformer structures as functions that map elements from a molecular graph directly to their 3D location in space. This formulation allows us to boil down the essence of structure prediction to learning a distribution over functions. Experimental results show that scaling up the model capacity leads to large gains in generalization performance without enforcing inductive biases like rotational equivariance. MCF represents an advance in extending diffusion models to handle complex scientific problems in a conceptually simple, scalable and effective manner.
-
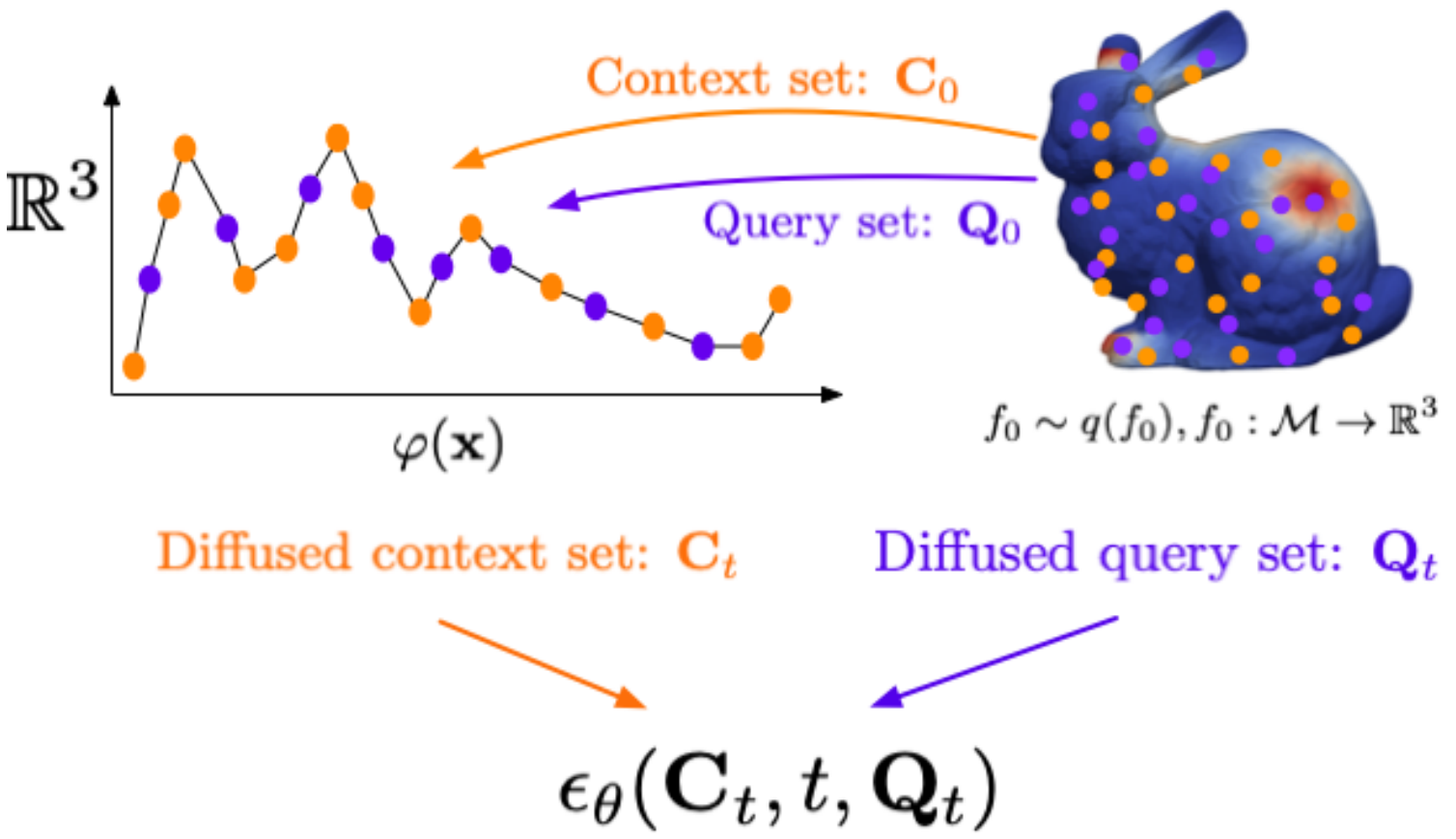 Manifold Diffusion FieldsAhmed A. Elhag, Yuyang Wang, Joshua Susskind, and Miguel Angel BautistaInternational Conference on Learning Representations (ICLR), 2024
Manifold Diffusion FieldsAhmed A. Elhag, Yuyang Wang, Joshua Susskind, and Miguel Angel BautistaInternational Conference on Learning Representations (ICLR), 2024We present Manifold Diffusion Fields (MDF), an approach that unlocks learning of diffusion models of data in general non-Euclidean geometries. Leveraging insights from spectral geometry analysis, we define an intrinsic coordinate system on the manifold via the eigen-functions of the Laplace-Beltrami Operator. MDF represents functions using an explicit parametrization formed by a set of multiple input-output pairs. Empirical results on multiple datasets and manifolds including challenging scientific problems like weather prediction or molecular conformation show that MDF can capture distributions of such functions with better diversity and fidelity than previous approaches.





2023
-
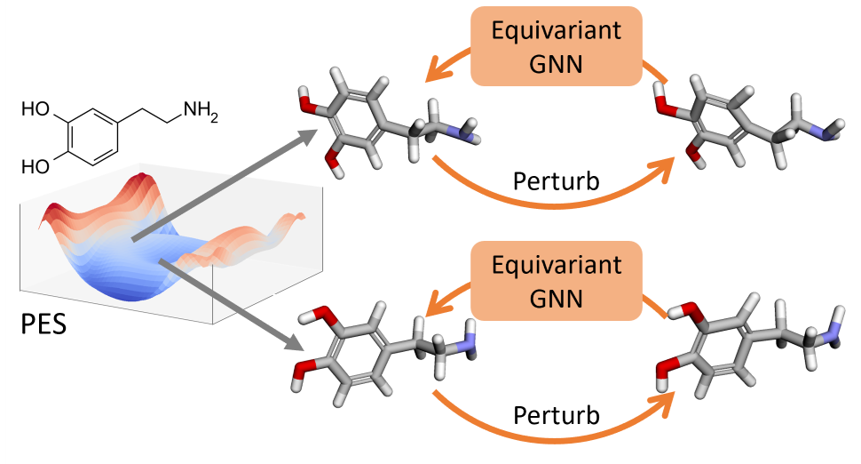 Denoise Pretraining on Nonequilibrium Molecules for Accurate and Transferable Neural PotentialsYuyang Wang, Changwen Xu, Zijie Li, and Amir Barati FarimaniJournal of Chemical Theory and Computation, 2023
Denoise Pretraining on Nonequilibrium Molecules for Accurate and Transferable Neural PotentialsYuyang Wang, Changwen Xu, Zijie Li, and Amir Barati FarimaniJournal of Chemical Theory and Computation, 2023Recent advances in equivariant graph neural networks (GNNs) have made deep learning amenable to developing fast surrogate models to expensive ab initio quantum mechanics (QM) approaches for molecular potential predictions. However, building accurate and transferable potential models using GNNs remains challenging, as the data is greatly limited by the expensive computational costs and level of theory of QM methods, especially for large and complex molecular systems. In this work, we propose denoise pretraining on nonequilibrium molecular conformations to achieve more accurate and transferable GNN potential predictions. Specifically, atomic coordinates of sampled nonequilibrium conformations are perturbed by random noises and GNNs are pretrained to denoise the perturbed molecular conformations which recovers the original coordinates. Rigorous experiments on multiple benchmarks reveal that pretraining significantly improves the accuracy of neural potentials. Furthermore, we show that the proposed pretraining approach is model-agnostic, as it improves the performance of different invariant and equivariant GNNs. Notably, our models pretrained on small molecules demonstrate remarkable transferability, improving performance when fine-tuned on diverse molecular systems, including different elements, charged molecules, biomolecules, and larger systems. These results highlight the potential for leveraging denoise pretraining approaches to build more generalizable neural potentials for complex molecular systems.
-
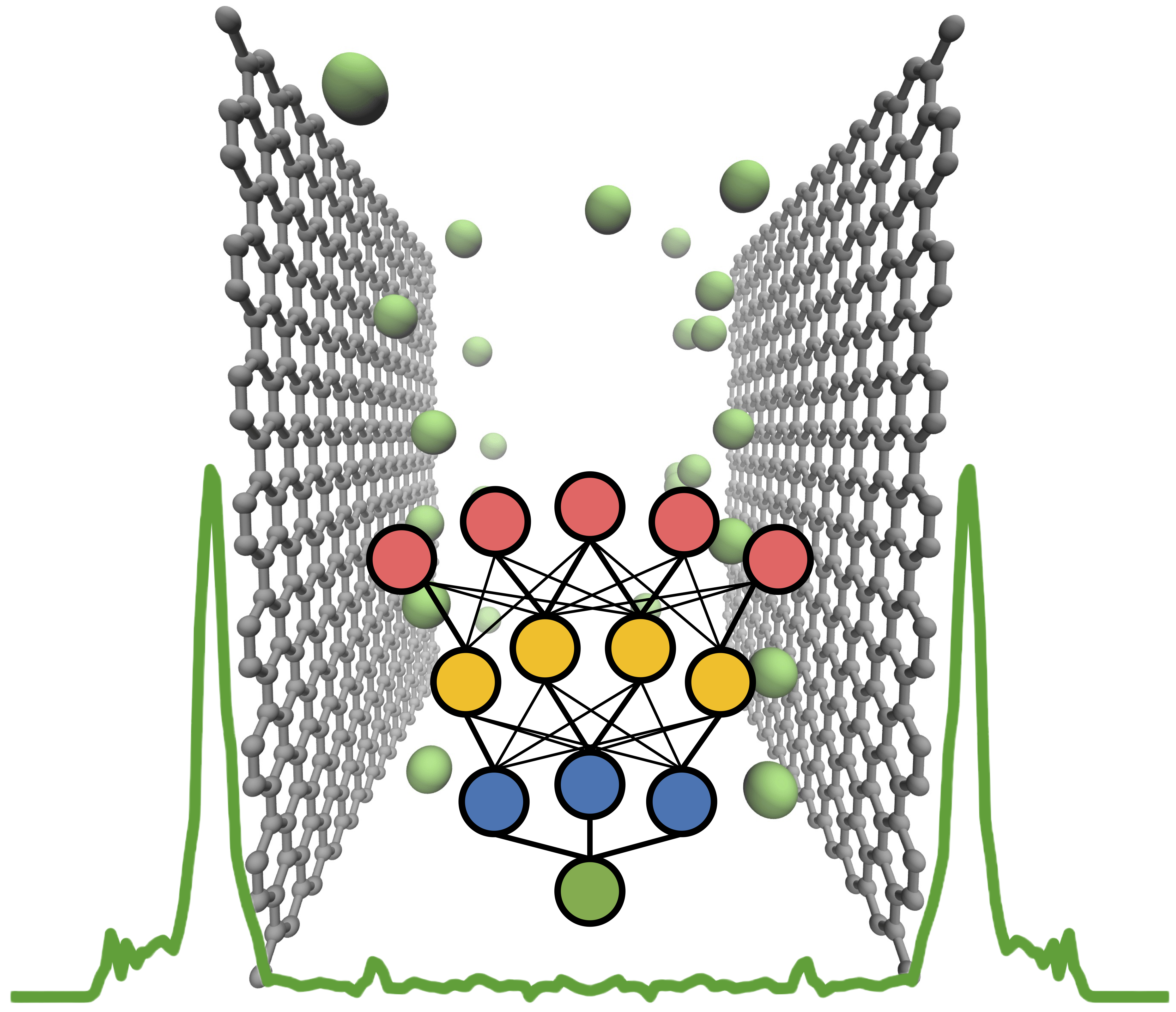 Neural Network Predicts Ion Concentration Profiles under NanoconfinementZhonglin Cao, Yuyang Wang, Cooper Lorsung, and Amir Barati FarimaniJournal of Chemical Physics, 2023
Neural Network Predicts Ion Concentration Profiles under NanoconfinementZhonglin Cao, Yuyang Wang, Cooper Lorsung, and Amir Barati FarimaniJournal of Chemical Physics, 2023Modeling the ion concentration profile in nanochannel plays an important role in understanding the electrical double layer and electroosmotic flow. Due to the non-negligible surface interaction and the effect of discrete solvent molecules, molecular dynamics (MD) simulation is often used as an essential tool to study the behavior of ions under nanoconfinement. Despite the accuracy of MD simulation in modeling nanoconfinement systems, it is computationally expensive. In this work, we propose neural network to predict ion concentration profiles in nanochannels with different configurations, including channel widths, ion molarity, and ion types. By modeling the ion concentration profile as a probability distribution, our neural network can serve as a much faster surrogate model for MD simulation with high accuracy. We further demonstrate the superior prediction accuracy of neural network over XGBoost. Lastly, we demonstrated that neural network is flexible in predicting ion concentration profiles with different bin sizes. Overall, our deep learning model is a fast, flexible, and accurate surrogate model to predict ion concentration profiles in nanoconfinement.
-
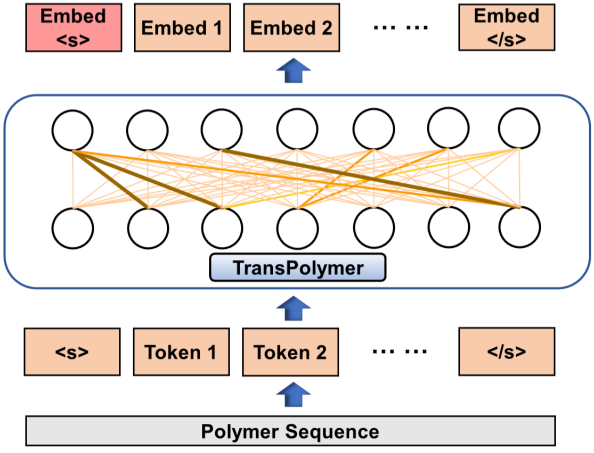 TransPolymer: a Transformer-based Language Model for Polymer Property PredictionsChangwen Xu, Yuyang Wang, and Amir Barati Farimaninpj Computational Materials, 2023
TransPolymer: a Transformer-based Language Model for Polymer Property PredictionsChangwen Xu, Yuyang Wang, and Amir Barati Farimaninpj Computational Materials, 2023Accurate and efficient prediction of polymer properties is of great significance in polymer design. Conventionally, expensive and time-consuming experiments or simulations are required to evaluate polymer functions. Recently, Transformer models, equipped with self-attention mechanisms, have exhibited superior performance in natural language processing. However, such methods have not been investigated in polymer sciences. Herein, we report TransPolymer, a Transformer-based language model for polymer property prediction. Our proposed polymer tokenizer with chemical awareness enables learning representations from polymer sequences. Rigorous experiments on ten polymer property prediction benchmarks demonstrate the superior performance of TransPolymer. Moreover, we show that TransPolymer benefits from pretraining on large unlabeled dataset via Masked Language Modeling. Experimental results further manifest the important role of self-attention in modeling polymer sequences. We highlight this model as a promising computational tool for promoting rational polymer design and understanding structure-property relationships from a data science view.
-
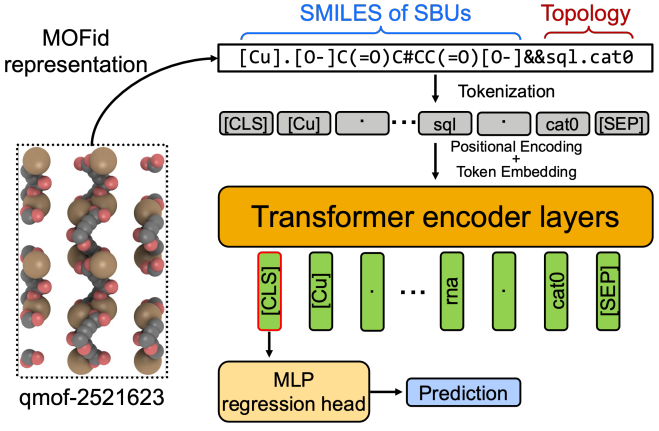 MOFormer: Self-Supervised Transformer Model for Metal-Organic Framework Property PredictionZhonglin Cao, Rishikesh Magar, Yuyang Wang, and Amir Barati FarimaniJournal of the American Chemical Society, 2023
MOFormer: Self-Supervised Transformer Model for Metal-Organic Framework Property PredictionZhonglin Cao, Rishikesh Magar, Yuyang Wang, and Amir Barati FarimaniJournal of the American Chemical Society, 2023Metal–organic frameworks (MOFs) are materials with a high degree of porosity that can be used for many applications. However, the chemical space of MOFs is enormous due to the large variety of possible combinations of building blocks and topology. Discovering the optimal MOFs for specific applications requires an efficient and accurate search over countless potential candidates. Previous high-throughput screening methods using computational simulations like DFT can be time-consuming. Such methods also require the 3D atomic structures of MOFs, which adds one extra step when evaluating hypothetical MOFs. In this work, we propose a structure-agnostic deep learning method based on the Transformer model, named as MOFormer, for property predictions of MOFs. MOFormer takes a text string representation of MOF (MOFid) as input, thus circumventing the need of obtaining the 3D structure of a hypothetical MOF and accelerating the screening process. By comparing to other descriptors such as Stoichiometric-120 and revised autocorrelations, we demonstrate that MOFormer can achieve state-of-the-art structure-agnostic prediction accuracy on all benchmarks. Furthermore, we introduce a self-supervised learning framework that pretrains the MOFormer via maximizing the cross-correlation between its structure-agnostic representations and structure-based representations of the crystal graph convolutional neural network (CGCNN) on >400k publicly available MOF data. Benchmarks show that pretraining improves the prediction accuracy of both models on various downstream prediction tasks. Furthermore, we revealed that MOFormer can be more data-efficient on quantum-chemical property prediction than structure-based CGCNN when training data is limited. Overall, MOFormer provides a novel perspective on efficient MOF property prediction using deep learning.
-
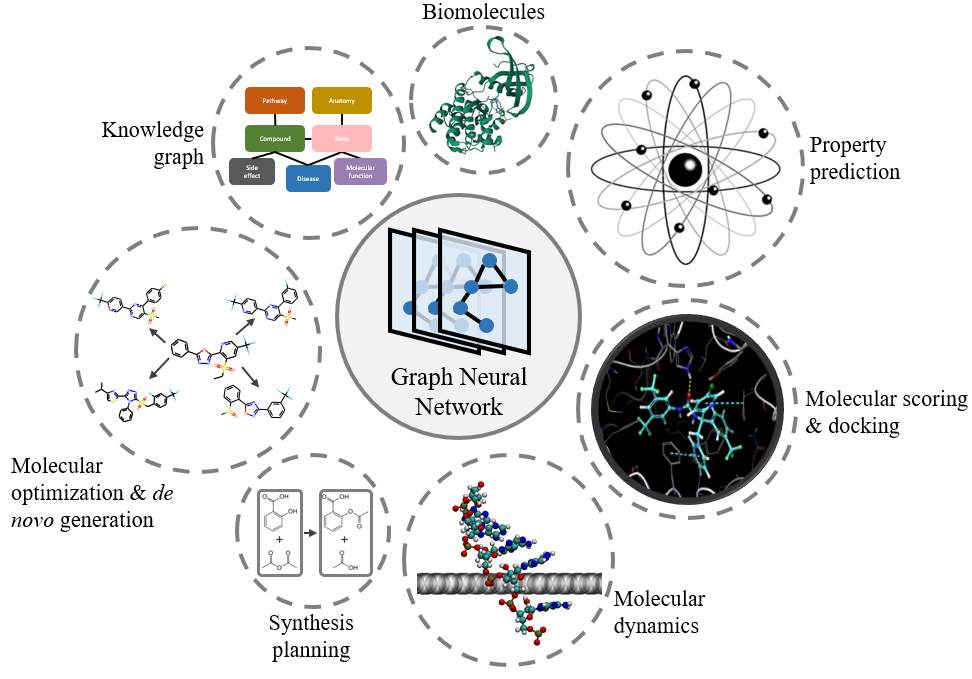 Graph Neural Networks for MoleculesYuyang Wang, Zijie Li, and Amir Barati FarimaniA chapter for book "Machine Learning in Molecular Sciences" published by Springer Nature, 2023
Graph Neural Networks for MoleculesYuyang Wang, Zijie Li, and Amir Barati FarimaniA chapter for book "Machine Learning in Molecular Sciences" published by Springer Nature, 2023Graph neural networks (GNNs), which are capable of learning representations from graphical data, are naturally suitable for modeling molecular systems. This review introduces GNNs and their various applications for small organic molecules. GNNs rely on message-passing operations, a generic yet powerful framework, to update node features iteratively. Many researches design GNN architectures to effectively learn topological information of 2D molecule graphs as well as geometric information of 3D molecular systems. GNNs have been implemented in a wide variety of molecular applications, including molecular property prediction, molecular scoring and docking, molecular optimization and de novo generation, molecular dynamics simulation, etc. Besides, the review also summarizes the recent development of self-supervised learning for molecules with GNNs.





2022
-
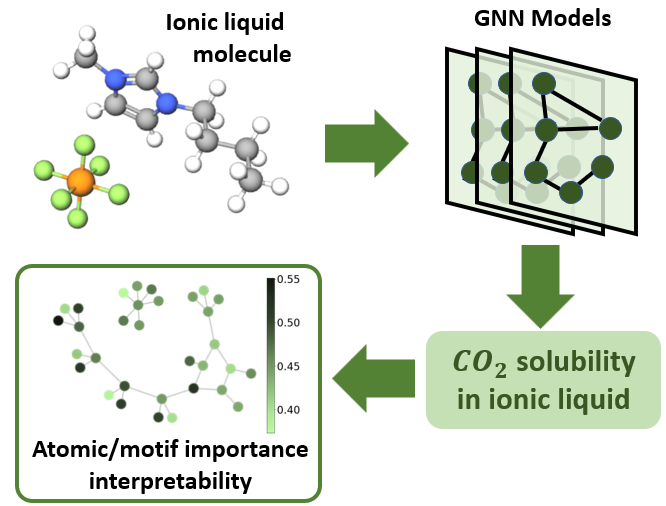 Predicting CO2 Absorption in Ionic Liquids with Molecular Descriptors and Explainable Graph Neural NetworksYue Jian, Yuyang Wang, and Amir Barati FarimaniACS Sustainable Chemistry & Engineering, 2022
Predicting CO2 Absorption in Ionic Liquids with Molecular Descriptors and Explainable Graph Neural NetworksYue Jian, Yuyang Wang, and Amir Barati FarimaniACS Sustainable Chemistry & Engineering, 2022Ionic Liquids (ILs) provide a promising solution for CO2 capture and storage to mitigate global warming. However, identifying and designing the high-capacity IL from the giant chemical space requires expensive, and exhaustive simulations and experiments. Machine learning (ML) can accelerate the process of searching for desirable ionic molecules through accurate and efficient property predictions in a data-driven manner. But existing descriptors and ML models for the ionic molecule suffer from the inefficient adaptation of molecular graph structure. Besides, few works have investigated the explainability of ML models to help understand the learned features that can guide the design of efficient ionic molecules. In this work, we develop both fingerprint-based ML models and Graph Neural Networks (GNNs) to predict the CO2 absorption in ILs. Fingerprint works on graph structure at the feature extraction stage, while GNNs directly handle molecule structure in both the feature extraction and model prediction stage. We show that our method outperforms previous ML models by reaching a high accuracy (MAE of 0.0137, R2 of 0.9884). Furthermore, we take the advantage of GNNs feature representation and develop a substructure-based explanation method that provides insight into how each chemical fragments within IL molecules contribute to the CO2 absorption prediction of ML models. We also show that our explanation result agrees with some ground truth from the theoretical reaction mechanism of CO2 absorption in ILs, which can advise on the design of novel and efficient functional ILs in the future.
-
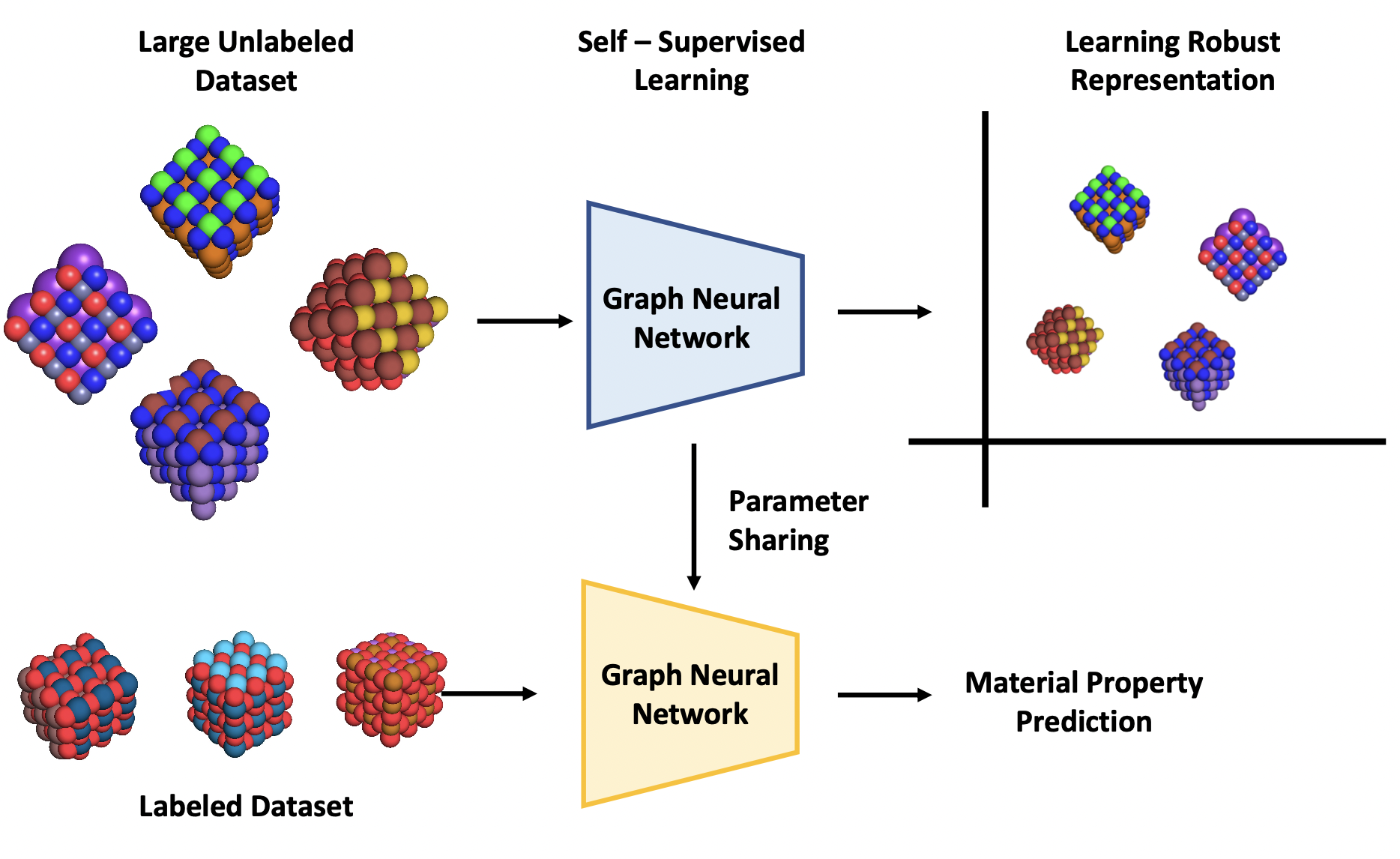 Crystal Twins: Self-supervised Learning for Crystalline Material Property PredictionRishikesh Magar, Yuyang Wang, and Amir Barati Farimaninpj Computational Materials, 2022
Crystal Twins: Self-supervised Learning for Crystalline Material Property PredictionRishikesh Magar, Yuyang Wang, and Amir Barati Farimaninpj Computational Materials, 2022Machine learning (ML) models have been widely successful in the prediction of material properties. However, large labeled datasets required for training accurate ML models are elusive and computationally expensive to generate. Recent advances in Self-Supervised Learning (SSL) frameworks capable of training ML models on unlabeled data have mitigated this problem and demonstrated superior performance in computer vision and natural language processing tasks. Drawing inspiration from the developments in SSL, we introduce Crystal Twins (CT): an SSL method for crystalline materials property prediction. Using a large unlabeled dataset, we pre-train a Graph Neural Network (GNN) by applying the redundancy reduction principle to the graph latent embeddings of augmented instances obtained from the same crystalline system. By sharing the pre-trained weights when fine-tuning the GNN for regression tasks, we significantly improve the performance for 7 challenging material property prediction benchmarks.
-
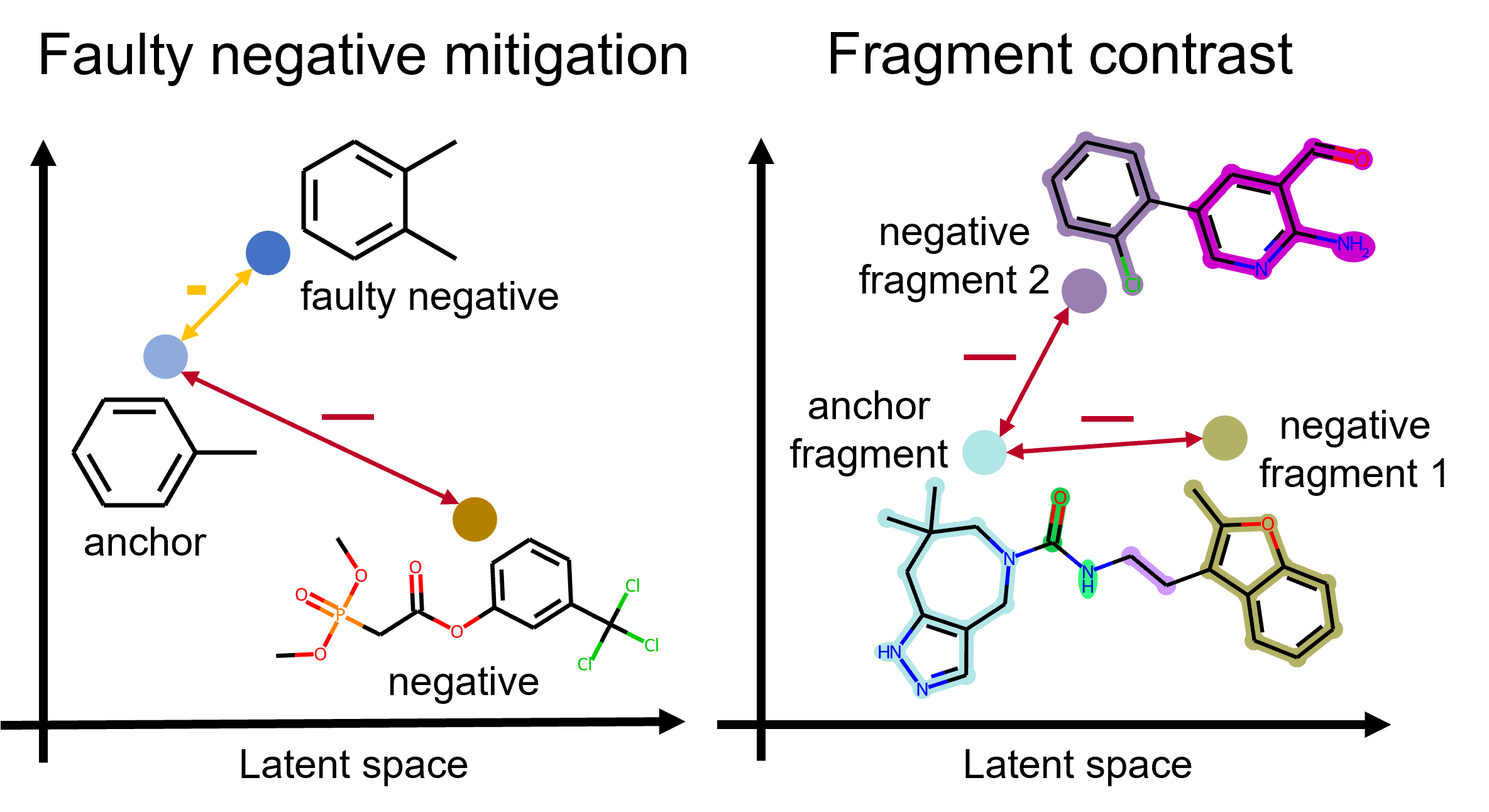 Improving Molecular Contrastive Learning via Faulty Negative Mitigation and Decomposed Fragment ContrastYuyang Wang, Rishikesh Magar, Chen Liang, and Amir Barati FarimaniJournal of Chemical Information and Modeling, 2022
Improving Molecular Contrastive Learning via Faulty Negative Mitigation and Decomposed Fragment ContrastYuyang Wang, Rishikesh Magar, Chen Liang, and Amir Barati FarimaniJournal of Chemical Information and Modeling, 2022Deep learning has been a prevalence in computational chemistry and widely implemented in molecule property predictions. Recently, self-supervised learning (SSL), especially contrastive learning (CL), gathers growing attention for the potential to learn molecular representations that generalize to the gigantic chemical space. Unlike supervised learning, SSL can directly leverage large unlabeled data, which greatly reduces the effort to acquire molecular property labels through costly and time-consuming simulations or experiments. However, most molecular SSL methods borrow the insights from the machine learning community but neglect the unique cheminformatics (e.g., molecular fingerprints) and multi-level graphical structures (e.g., functional groups) of molecules. In this work, we propose iMolCLR: improvement of Molecular Contrastive Learning of Representations with graph neural networks (GNNs) in two aspects, (1) mitigating faulty negative contrastive instances via considering cheminformatics similarities between molecule pairs; (2) fragment-level contrasting between intra- and inter-molecule substructures decomposed from molecules. Experiments have shown that the proposed strategies significantly improve the performance of GNN models on various challenging molecular property predictions. In comparison to the previous CL framework, iMolCLR demonstrates an averaged 1.2% improvement of ROC-AUC on 8 classification benchmarks and an averaged 10.1% decrease of the error on 6 regression benchmarks. On most benchmarks, the generic GNN pre-trained by iMolCLR rivals or even surpasses supervised learning models with sophisticated architecture designs and engineered features. Further investigations demonstrate that representations learned through iMolCLR intrinsically embed scaffolds and functional groups that can reason molecule similarities.
-
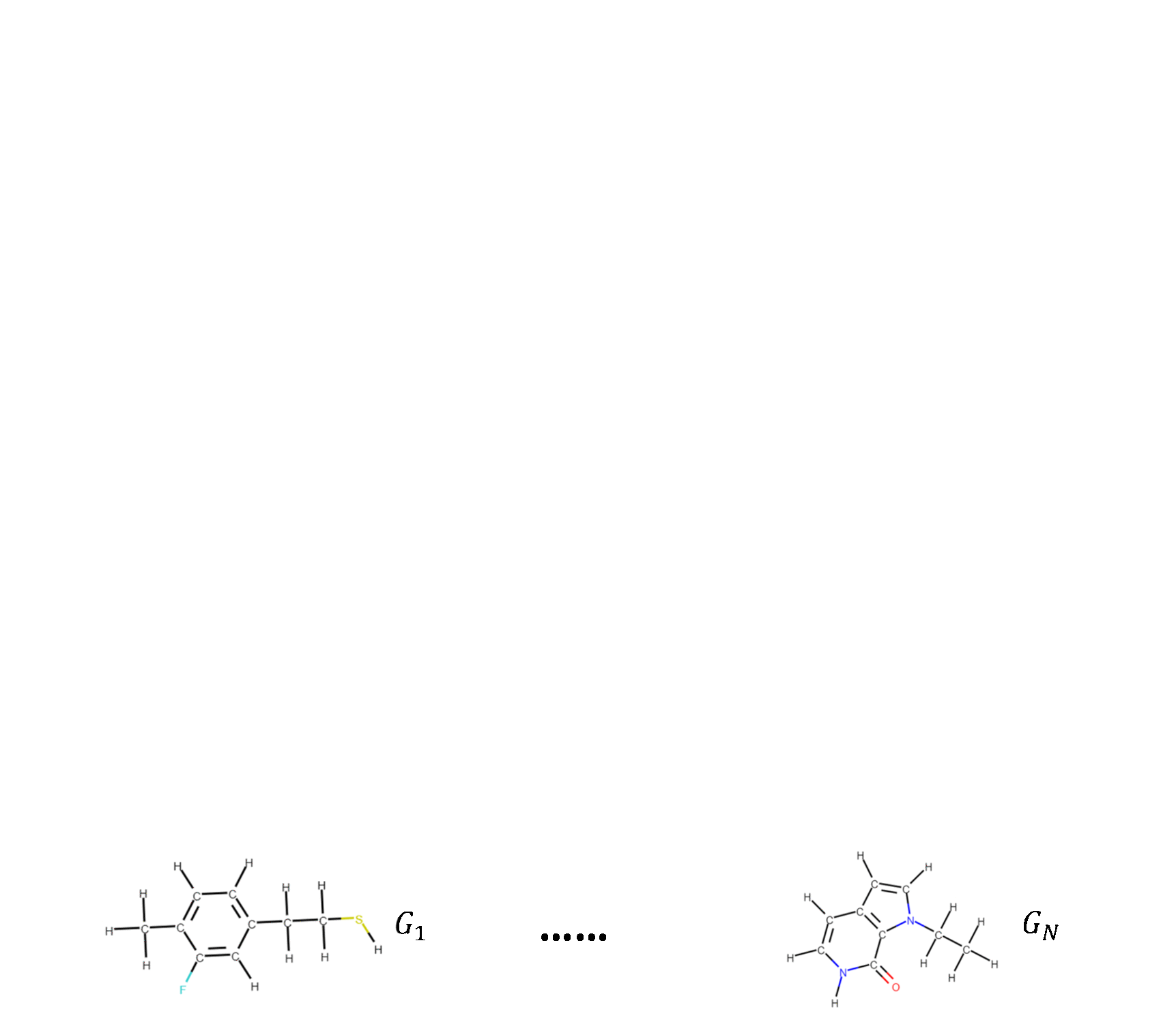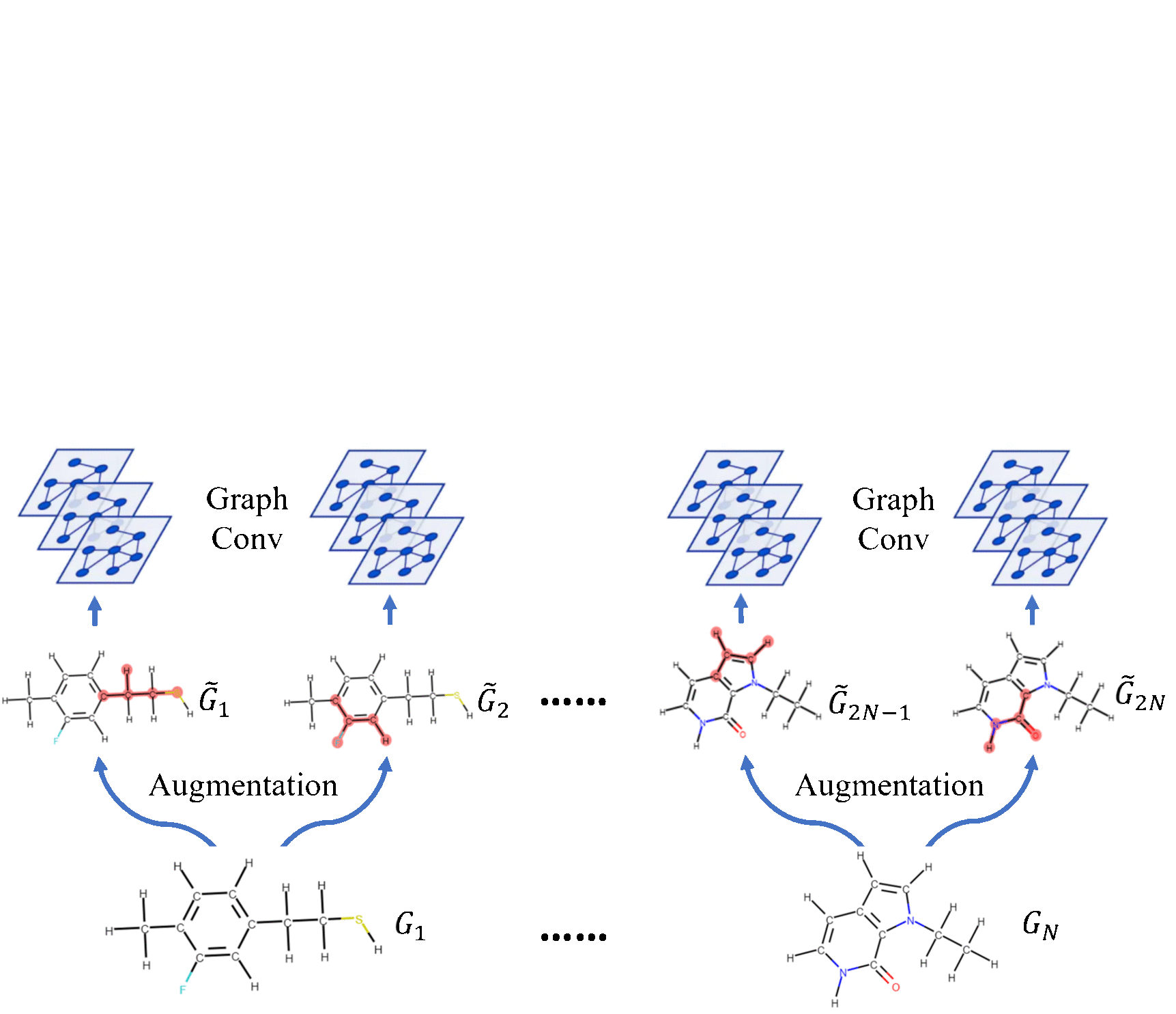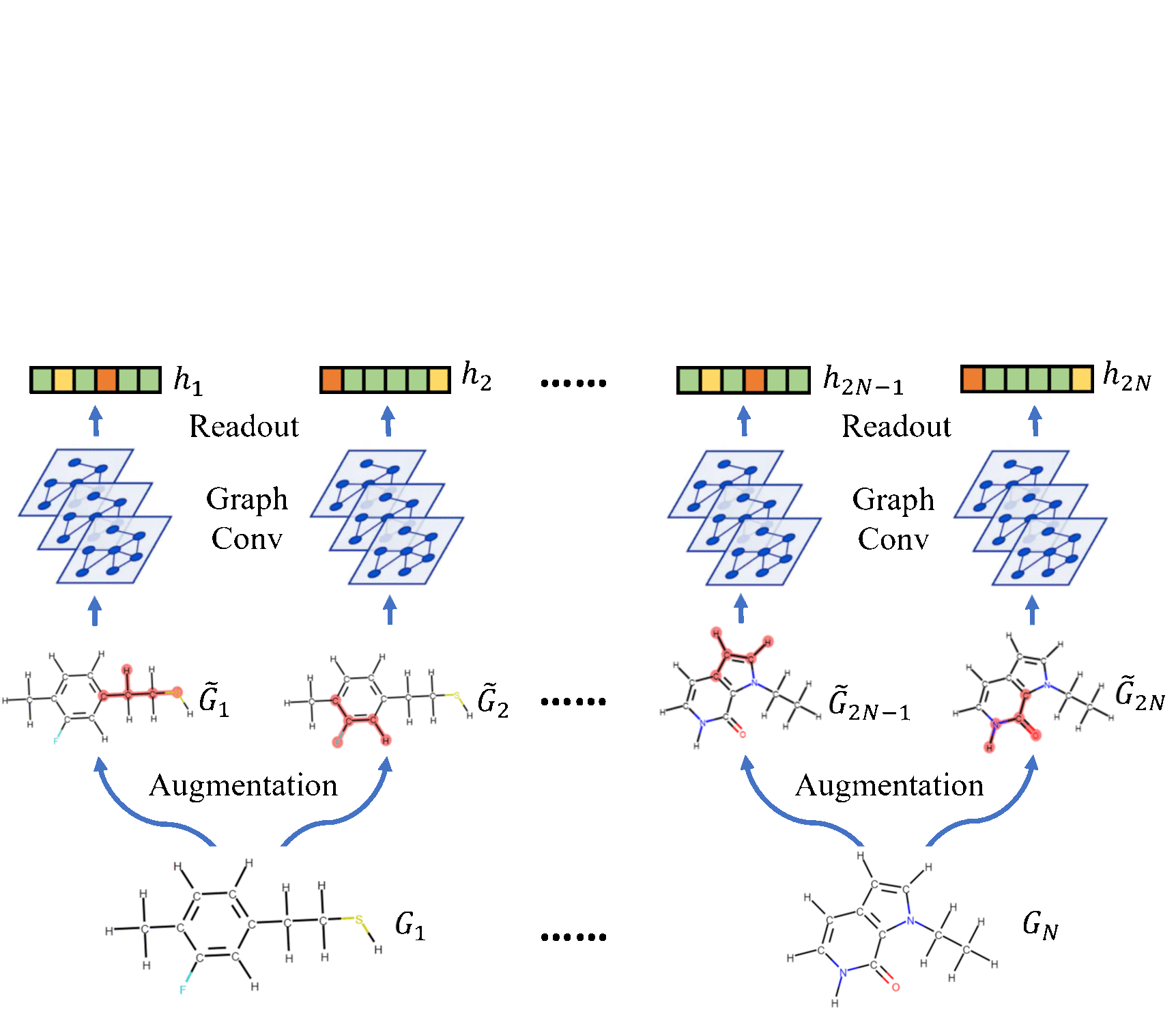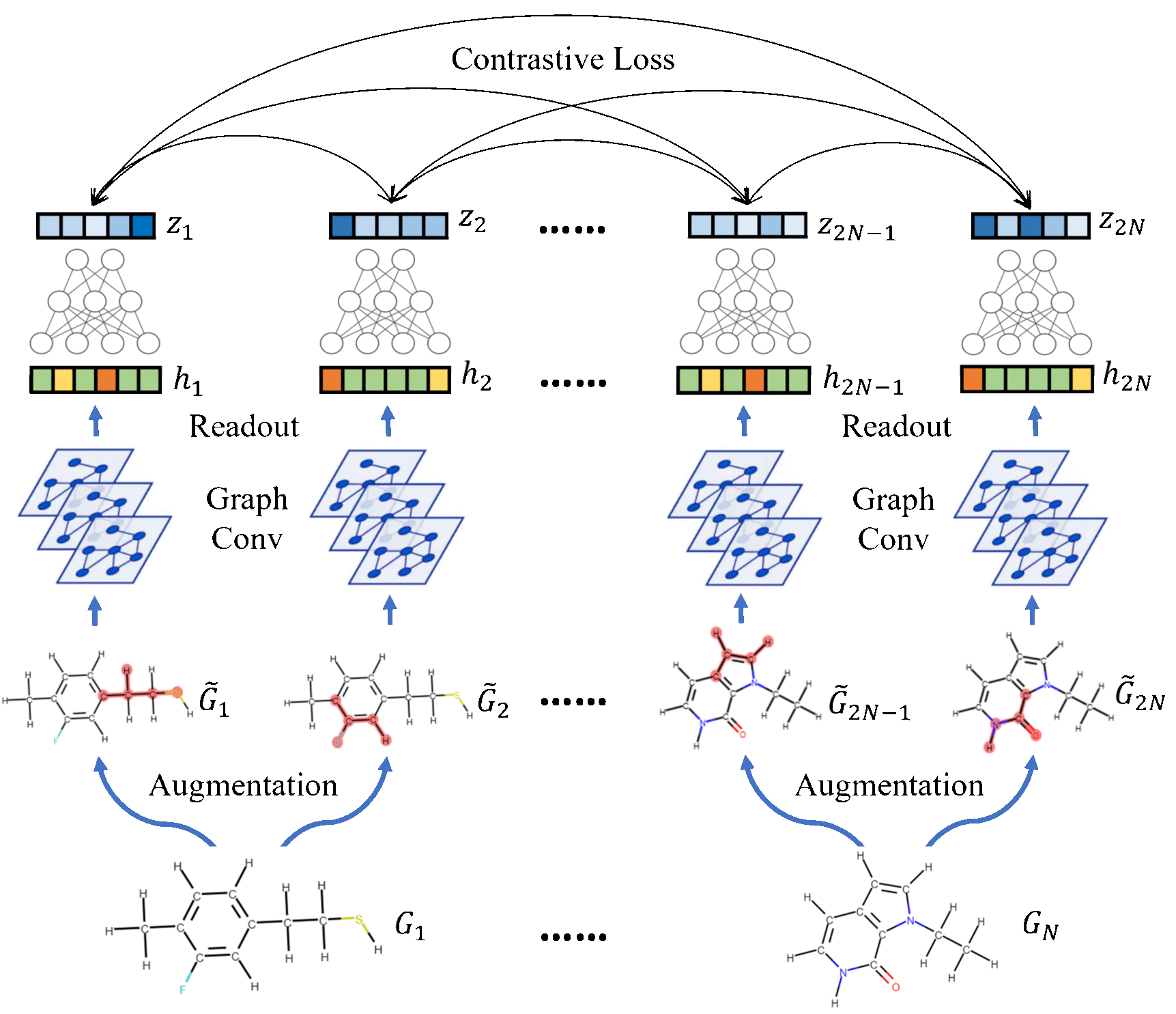 Molecular Contrastive Learning of Representations via Graph Neural NetworksYuyang Wang, Jianren Wang, Zhonglin Cao, and Amir Barati FarimaniNature Machine Intelligence, 2022
Molecular Contrastive Learning of Representations via Graph Neural NetworksYuyang Wang, Jianren Wang, Zhonglin Cao, and Amir Barati FarimaniNature Machine Intelligence, 2022Molecular machine learning bears promise for efficient molecular property prediction and drug discovery. However, labelled molecule data can be expensive and time consuming to acquire. Due to the limited labelled data, it is a great challenge for supervised-learning machine learning models to generalize to the giant chemical space. Here we present MolCLR (Molecular Contrastive Learning of Representations via Graph Neural Networks), a self-supervised learning framework that leverages large unlabelled data ( 10 million unique molecules). In MolCLR pre-training, we build molecule graphs and develop graph-neural-network encoders to learn differentiable representations. Three molecule graph augmentations are proposed: atom masking, bond deletion and subgraph removal. A contrastive estimator maximizes the agreement of augmentations from the same molecule while minimizing the agreement of different molecules. Experiments show that our contrastive learning framework significantly improves the performance of graph-neural-network encoders on various molecular property benchmarks including both classification and regression tasks. Benefiting from pre-training on the large unlabelled database, MolCLR even achieves state of the art on several challenging benchmarks after fine-tuning. In addition, further investigations demonstrate that MolCLR learns to embed molecules into representations that can distinguish chemically reasonable molecular similarities.
-
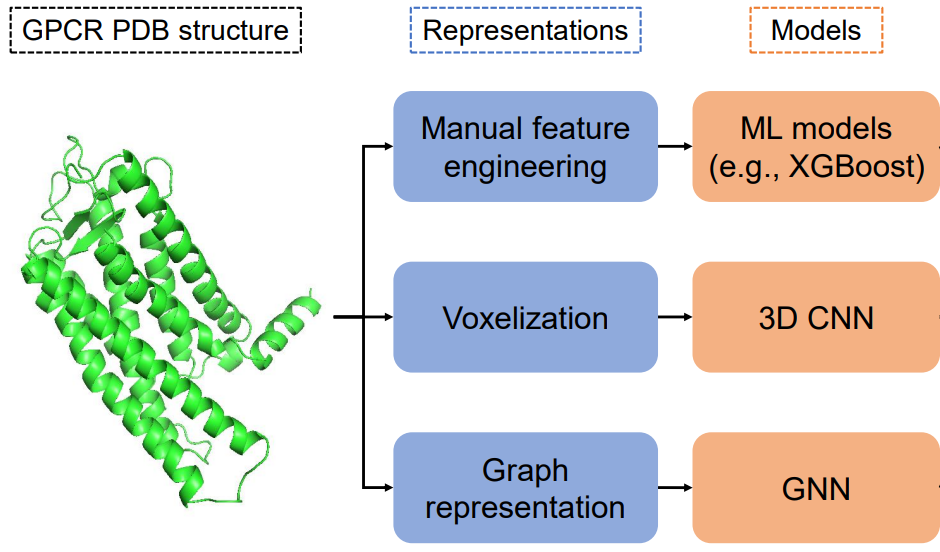 Prediction of GPCR activity using Deep LearningPrakarsh Yadav, Parisa Mollaei, Zhonglin Cao, Yuyang Wang, and Amir Barati FarimaniComputational and Structural Biotechnology Journal, 2022
Prediction of GPCR activity using Deep LearningPrakarsh Yadav, Parisa Mollaei, Zhonglin Cao, Yuyang Wang, and Amir Barati FarimaniComputational and Structural Biotechnology Journal, 2022GPCRs are the target for one-third of the FDA-approved drugs, however; the development of new drug molecules targeting GPCRs is limited by the lack of mechanistic understanding of the GPCR structure-activity-function relationship. To modulate the GPCR activity with highly specific drugs and minimal side-effects, it is necessary to quantitatively describe the important structural features in the GPCR and correlate them to the activation state of GPCR. In this study, we developed 3 ML approaches to predict the conformation state of GPCR proteins. Additionally, we predict the activity level of GPCRs based on their structure. We leverage the unique advantages of each of the 3 ML approach, interpretability of XGBoost, minimal feature engineering for 3D convolutional neural network, and graph representation of protein structure for graph neural network. By using these ML approaches, we are able to predict the GPCRs activation state with high accuracy (91%-95%) and also predict the activation state of GPCRs with low error (MAE of 7.15-10.58). Furthermore, the interpretation of the ML approaches allow us to determine the importance of each of the features in distinguishing between the GPCRs conformations.
-
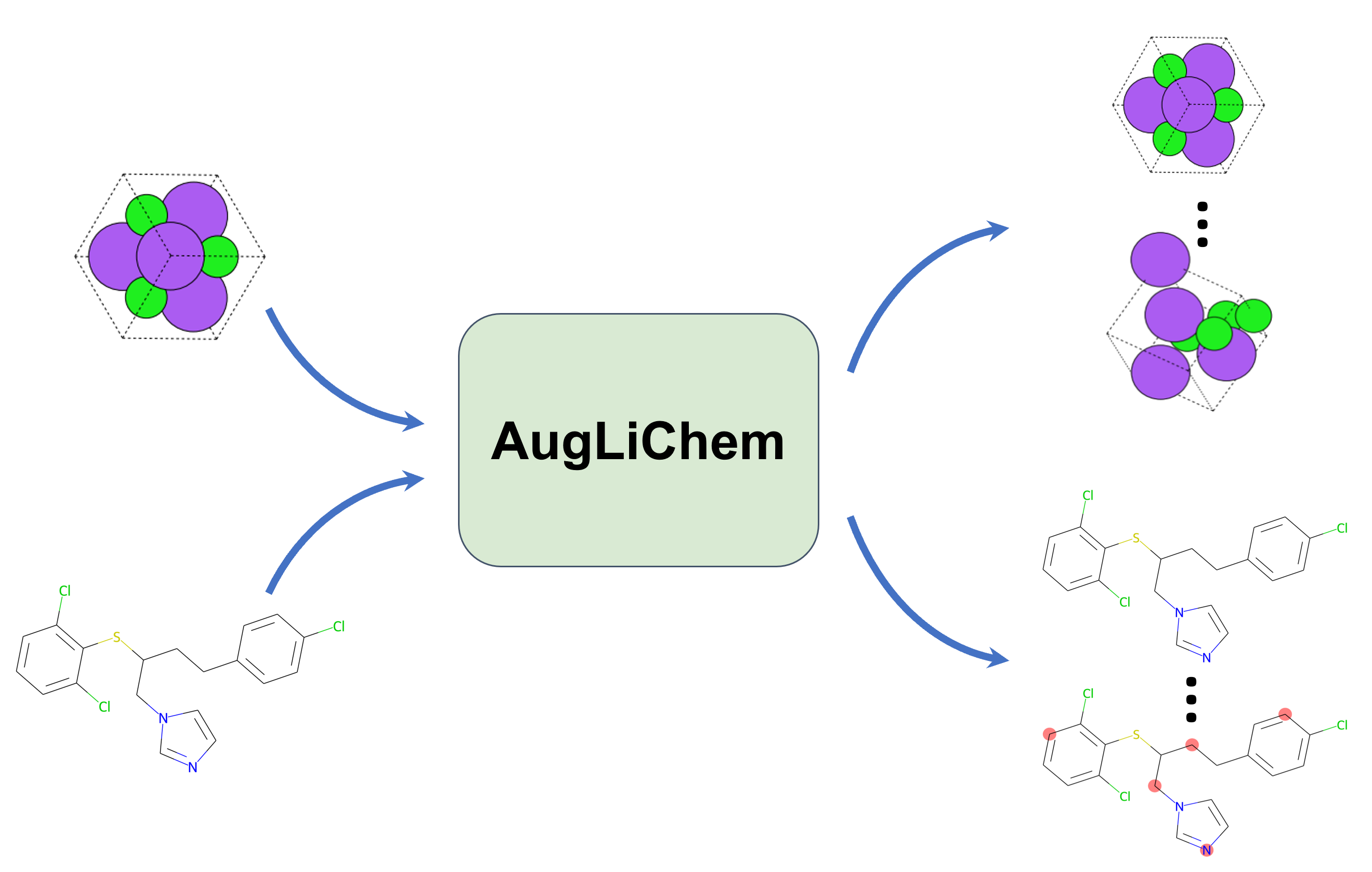 AugLiChem: Data Augmentation Library of Chemical Structures for Machine LearningRishikesh Magar*, Yuyang Wang*, Cooper Lorsung*, Chen Liang, Hariharan Ramasubramanian, Peiyuan Li, and Amir Barati FarimaniMachine Learning: Science and Technology, 2022
AugLiChem: Data Augmentation Library of Chemical Structures for Machine LearningRishikesh Magar*, Yuyang Wang*, Cooper Lorsung*, Chen Liang, Hariharan Ramasubramanian, Peiyuan Li, and Amir Barati FarimaniMachine Learning: Science and Technology, 2022Machine learning (ML) has demonstrated the promise for accurate and efficient property prediction of molecules and crystalline materials. To develop highly accurate ML models for chemical structure property prediction, datasets with sufficient samples are required. However, obtaining clean and sufficient data of chemical properties can be expensive and time-consuming, which greatly limits the performance of ML models. Inspired by the success of data augmentations in computer vision and natural language processing, we developed AugLiChem: the data augmentation library for chemical structures. Augmentation methods for both crystalline systems and molecules are introduced, which can be utilized for fingerprint-based ML models and Graph Neural Networks (GNNs). We show that using our augmentation strategies significantly improves the performance of ML models, especially when using GNNs. In addition, the augmentations that we developed can be used as a direct plug-in module during training and have demonstrated the effectiveness when implemented with different GNN models through the AugliChem library. The Python-based package for our implementation of Auglichem: Data augmentation library for chemical structures, is publicly available at: https://github.com/BaratiLab/AugLiChem.






2021
-
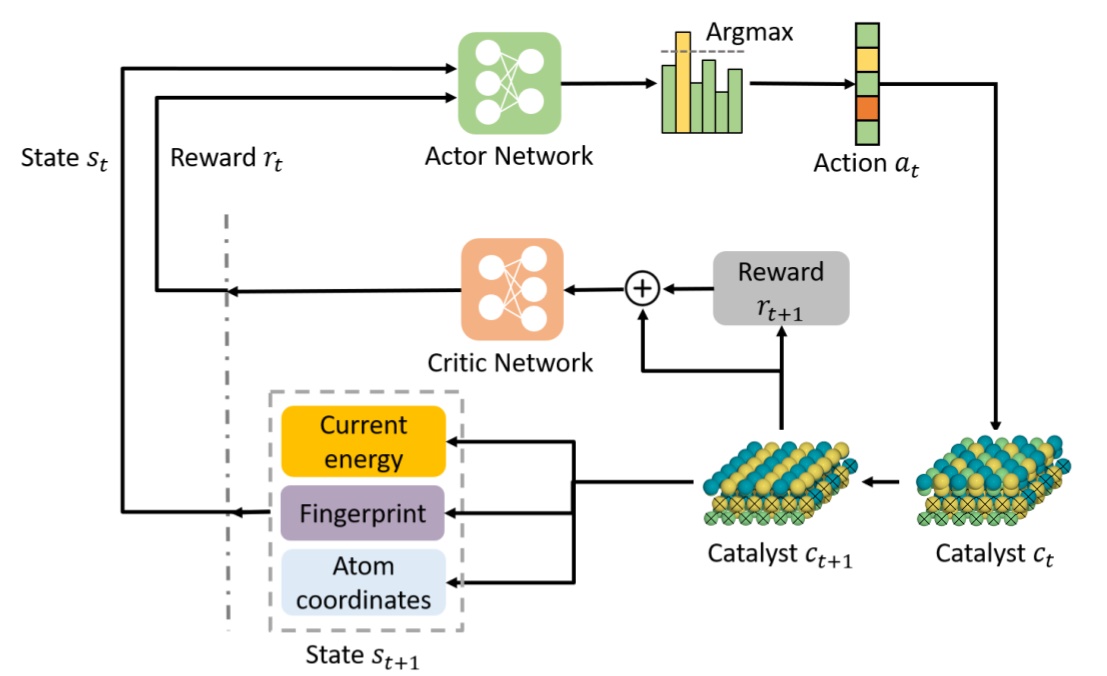 Deep Reinforcement Learning for Predicting Kinetic Pathways to Surface Reconstruction in a Ternary Alloy.Junwoong Yoon, Zhonglin Cao, Rajesh Raju, Yuyang Wang, Robert Burnley, Andrew J Gellman, Amir Barati Farimani, and Zachary W UlissiMachine Learning: Science and Technology, 2021
Deep Reinforcement Learning for Predicting Kinetic Pathways to Surface Reconstruction in a Ternary Alloy.Junwoong Yoon, Zhonglin Cao, Rajesh Raju, Yuyang Wang, Robert Burnley, Andrew J Gellman, Amir Barati Farimani, and Zachary W UlissiMachine Learning: Science and Technology, 2021The majority of computational catalyst design focuses on the screening of material components and alloy composition to optimize selectivity and activity for a given reaction. However, predicting the metastability of the alloy catalyst surface at realistic operating conditions requires an extensive sampling of possible surface reconstructions and their associated kinetic pathways. We present CatGym, a deep reinforcement learning (DRL) environment for predicting the thermal surface reconstruction pathways and their associated kinetic barriers in crystalline solids under reaction conditions. The DRL agent iteratively changes the positions of atoms in the near-surface region to generate kinetic pathways to accessible local minima involving changes in the surface compositions. We showcase our agent by predicting the surface reconstruction pathways of a ternary Ni3Pd3Au2(111) alloy catalyst. Our results show that the DRL agent can not only explore more diverse surface compositions than the conventional minima hopping method, but also generate the kinetic surface reconstruction pathways. We further demonstrate that the kinetic pathway to a global minimum energy surface composition and its associated transition state predicted by our agent is in good agreement with the minimum energy path predicted by nudged elastic band calculations.
-
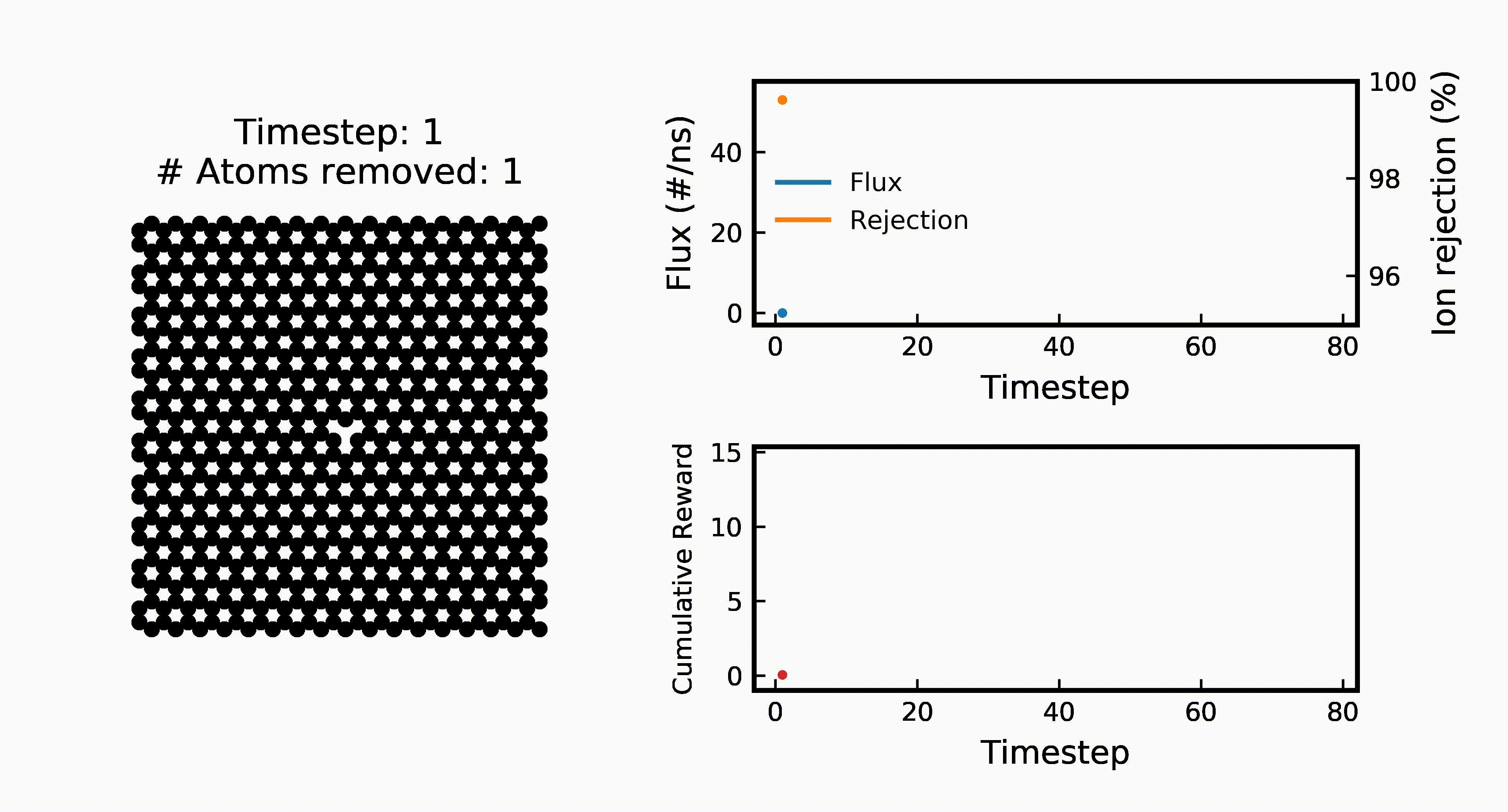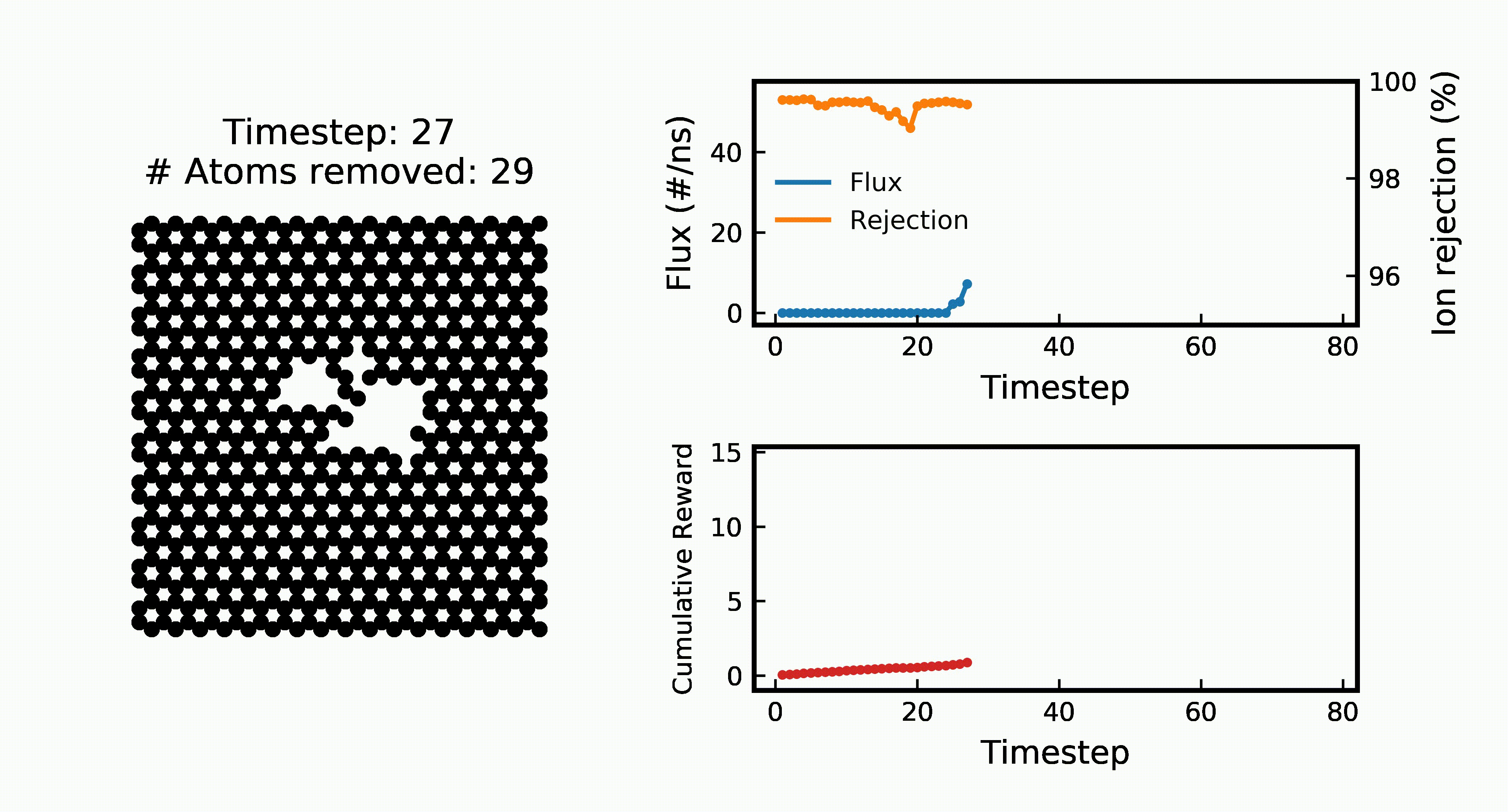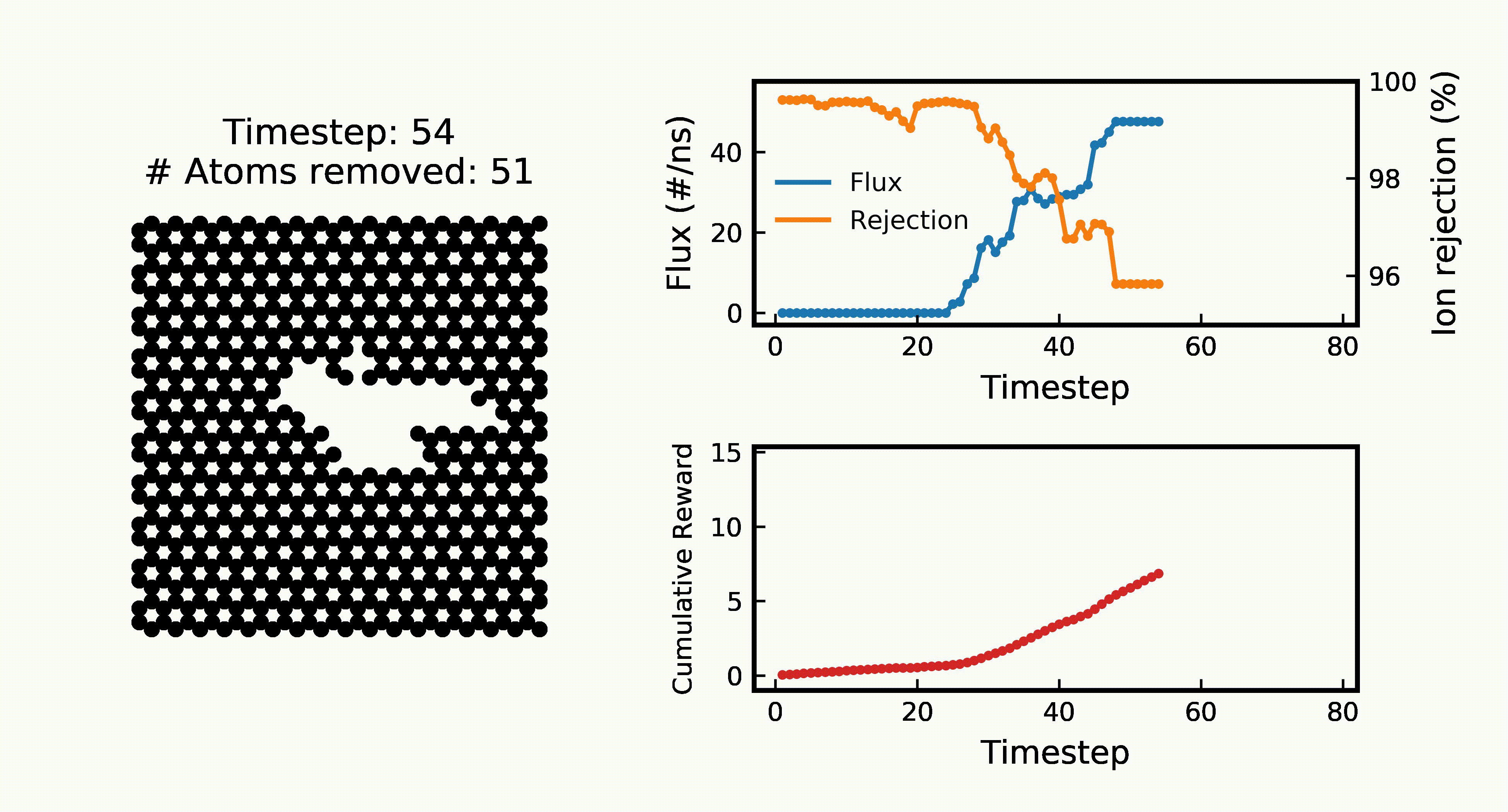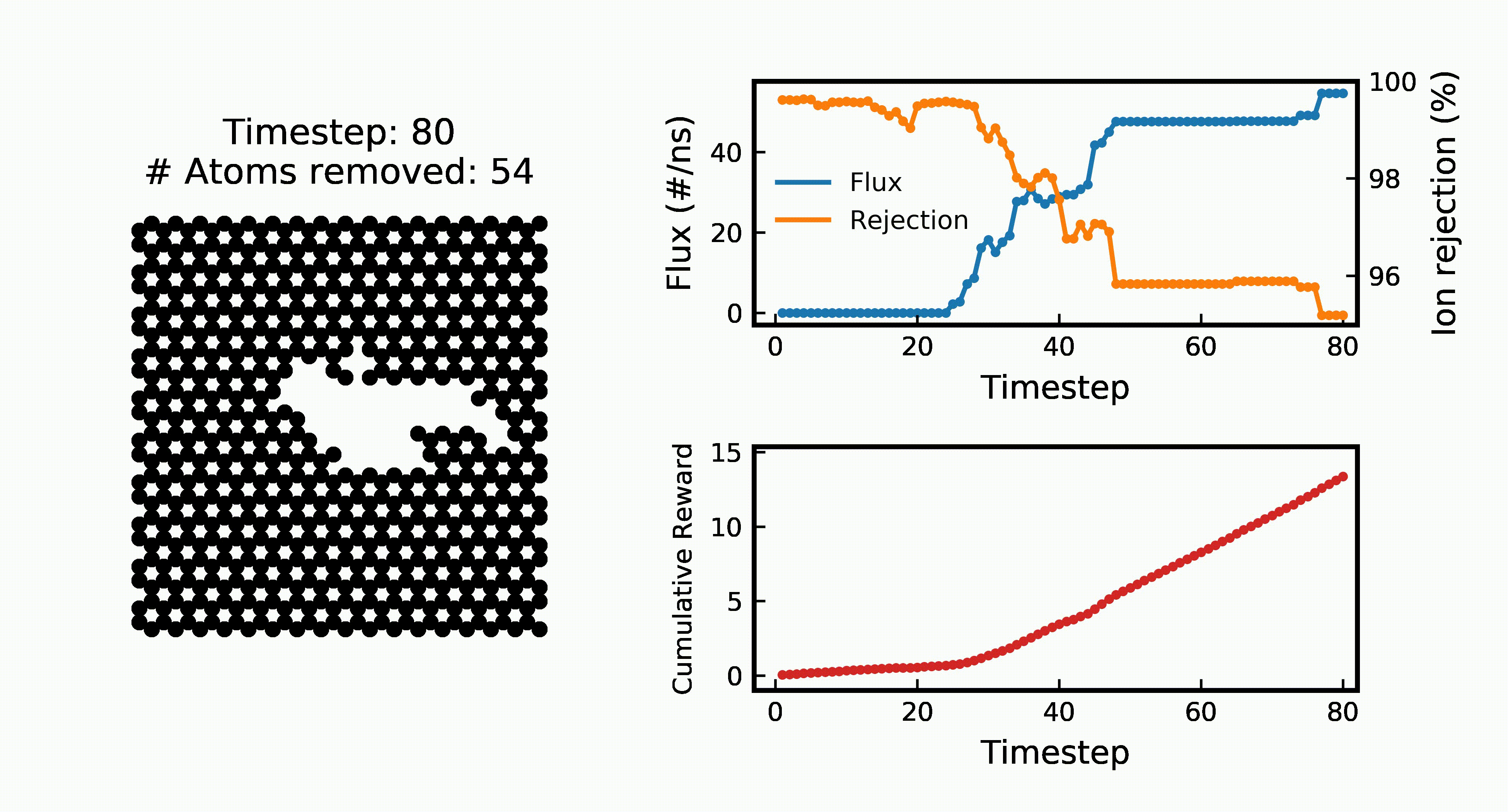 Efficient Water Desalination with Graphene Nanopores Obtained using Artificial IntelligenceYuyang Wang*, Zhonglin Cao*, and Amir Barati Farimaninpj 2D Materials and Applications, 2021
Efficient Water Desalination with Graphene Nanopores Obtained using Artificial IntelligenceYuyang Wang*, Zhonglin Cao*, and Amir Barati Farimaninpj 2D Materials and Applications, 2021Two-dimensional nanomaterials, such as graphene, have been extensively studied because of their outstanding physical properties. Structure and topology of nanopores on such materials can be important for their performances in real-world engineering applications, like water desalination. However, discovering the most efficient nanopores often involves a very large number of experiments or simulations that are expensive and time-consuming. In this work, we propose a data-driven artificial intelligence (AI) framework for discovering the most efficient graphene nanopore for water desalination. Via a combination of deep reinforcement learning (DRL) and convolutional neural network (CNN), we are able to rapidly create and screen thousands of graphene nanopores and select the most energy-efficient ones. Molecular dynamics (MD) simulations on promising AI-created graphene nanopores show that they have higher water flux while maintaining rival ion rejection rate compared to the normal circular nanopores. Irregular shape with rough edges geometry of AI-created pores is found to be the key factor for their high water desalination performance. Ultimately, this study shows that AI can be a powerful tool for nanomaterial design and screening.
-
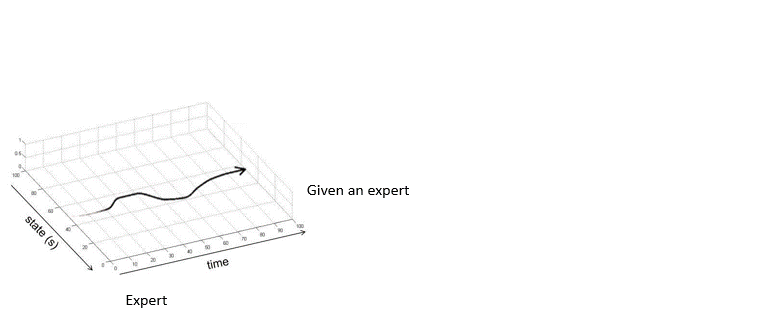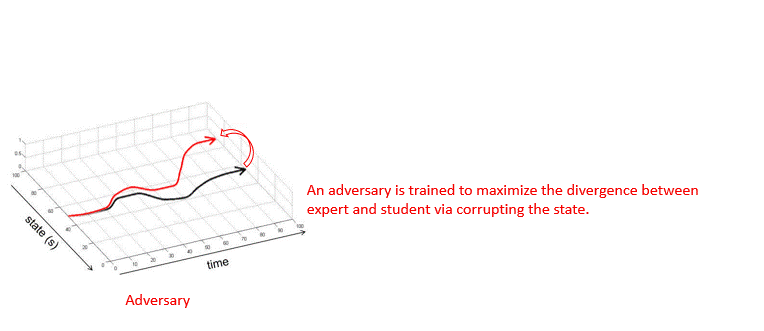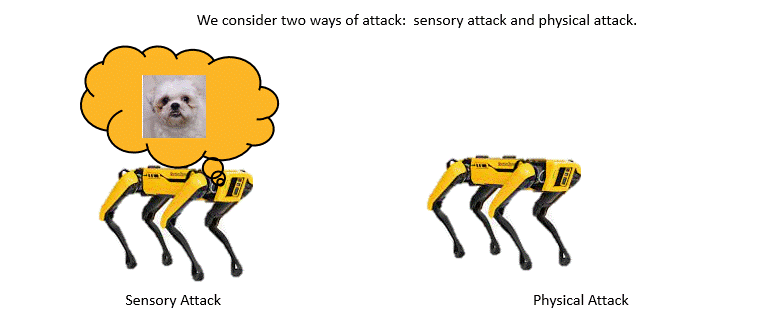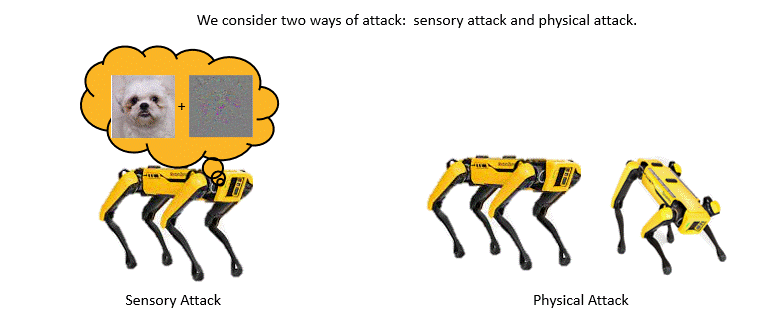 Adversarially Robust Imitation LearningJianren Wang, Ziwen Zhuang, Yuyang Wang, and Hang ZhaoIn 5th Annual Conference on Robot Learning, 2021
Adversarially Robust Imitation LearningJianren Wang, Ziwen Zhuang, Yuyang Wang, and Hang ZhaoIn 5th Annual Conference on Robot Learning, 2021Modern imitation learning (IL) utilizes deep neural networks (DNNs) as function approximators to mimic the policy of the expert demonstrations. However, DNNs can be easily fooled by subtle noise added to the input, which is even non-detectable by humans. This makes the learned agent vulnerable to attacks, especially in IL where agents can struggle to recover from the errors. In such light, we propose a sound Adversarially Robust Imitation Learning (ARIL) method. In our setting, an agent and an adversary are trained alternatively. The former with adversarially attacked input at each timestep mimics the behavior of an online expert and the latter learns to add perturbations on the states by forcing the learned agent to fail on choosing the right decisions. We theoretically prove that ARIL can achieve adversarial robustness and evaluate ARIL on multiple benchmarks from DM Control Suite. The result reveals that our method (ARIL) achieves better robustness compare with other imitation learning methods under both sensory attack and physical attack.


